ISSN: 0970-938X (Print) | 0976-1683 (Electronic)
Biomedical Research
An International Journal of Medical Sciences
Research Article - Biomedical Research (2021) Volume 32, Issue 1
In silico mechanistic intervention of medicinal plants derived inhibitors against ABL kinase targeting cervical cancer: A novel approach
1Department of Bioinformatics, Alagappa University, Karaikudi, Tamil Nadu, India
2Department of Animal Health and Management, Alagappa University, Karaikudi, Tamil Nadu, India
- Corresponding Author:
- Vidhyavathi RM
Department of Bioinformatics
Science Campus
Alagappa University
Karaikudi
Tamil Nadu
India
Accepted on December 07, 2020
Aim: The present investigation aimed to evaluate potential inhibitors against ABL Kinase. Background: Cervical Cancer. Cervical cancer is a chronic disorder and 4th most recurring worldwide cancer occurring in women. It has been estimated that 5700,000 emerging cares were diagnosed in 2018. However, it still remains unresolved and immediately requires a powerful lead compound for its detainment. Human Papilloma Viruses (HPV) is well known to be the causative agent for the major sections of the cervical cancer cases. ABL Kinase is a protein and represents a close associate with activities of HPV and widely influencing cancer therapy. It involved in directing the vital functions of cells covering, multiplicity of cells, differentiation, offering feedback to oxidative stress and its survival. Objective: To find a novel inhibitor for Kinase targeting Cervical Cancer from medicinal plants. Method: Therefore, an effort has been attempted to identify lead compounds from 20 selected medicinal plants against ABL Kinase protein, implicating cervical cancer. ABL Kinase 3D structural analysis was performed to point out highly powerful inhibitors against them using virtual screening. Results: The results showed that the identified lead compound Apigenin Dimethyl ether was found to be more effective and are abundant in Andrographis paniculata species. The stability of the lead compounds was further examined using molecular simulation and resulting in a stability of the complex was well established. Based on the protein-ligand binding free energy, chemical reactions of the lead compounds were also determined for its suitability. An analysis of hit compounds profile was investigated for its suitability in consumption ability, safety and for better pharmaceutical marketing. Moreover, ADME properties of hit compounds were also predicted and were found within the limit of Lipinski-5 rules. Conclusion: This study concluded that based on the outcomes of activation energy Apigenin Dimethyl ether has proven to be a powerful inhibitor against ABL Kinase and can be considered for the successful treatment of cervical cancer in clinical settings.
Keywords
Molecular dynamics, Docking, ABL Kinase inhibitors, Cervical cancer, Virtual screening, Andrographis paniculata.
Introduction
According to the recent WHO report (2020), Cervical cancer is ranked 4th most commonly incidence cancer in women and is accounted for 8th place in the list of most occurring overall cancer in the world. Cervical cancer caused inevitably, 275100 deaths in every year [1]. Virtually, diagnosed all cervical malignant growths are closely linked with infection of Human Papilloma Virus (HPV). Studies have reported that 2 to 20 % of women in the world are found with detectable signs of HPV viral DNA in their cervix [2]. However, a major section of women with HPV do not develop cervical cancer, in fact, it requires other external factors for developing malignant, after HPV infections [3]. HPVs are small in size, unenveloped, with double stranded DNA and parasitic nature similar to other viruses.
Current clinical therapies, existing in force, such as chemotherapy, and radiotherapy conferred unsuccessful outcomes due to the recurrence of invasion and metastasis, resulting in patients with disappointment in the treatments [4]. Moreover, current therapies, have quite limitations in treating certain unsure phases of the ailment and conferred hectic worries while using radiotherapy [5].
ABL kinase is a protein, in early, invented in Abelson murine leukemia virus 1, and was recognised as an oncogene, involved and closely associated with a process of chromosome translocations, resulting in cell transformation [6-8]. However, the activation of ABL 1 and ABL 2 kinases in strong tumours remains unclear. It has been reported that the potential of ABL kinase to the strong malignant was found to be uncertain during chromosome translocations phenomenon [9]. However, it has been known to act as a driven force by upgrading ABL 1 and ABL 2 due to the activities of oncogene tri kinases, chemokine receptors, intolerable stress, and disabling unresponsive proteins [10].
By considering important functions of ABL kinase in directing cell transformation, covering cell extremity, invasion, amplification and persistence, in addition, FDA approved popular drugs such as imatinib and dasatinib currently used for treating strong tumors have led to unsuccessful results with undesirable side effects. Moreover, several investigations have demonstrated that there is a need to develop tyrosine kinase inhibitors to treat various chronic diseases [11-13]. Limited studies were published on the development of ABL kinase inhibitors with care to discriminate biomarkers for determined classified [14].
In recent time, a mountain of evidence deciphered that few bioactive ingredients of medicinal plants derived have been exercised with anticancer potentials and used for treating various ailments. Therefore, in the present investigation, the main objective was to find a strong inhibitor against ABL kinase protein using mechanistic docking strategy with plant-based ligands, based on their free energy, and interaction ability by employing virtual screening.
Materials and Methods
The present investigation involved mechanistic intervention and analysis of docking and screening of physico chemical characteristics of selected medicinal plants bioactive compounds by employing DFT, MMGBSA, and ADME, followed by molecular simulations were executed using Maestro Version 18-4.
Preparation of protein and ligands
Crystalline structure of ABL kinase protein (ID: 2HZ1) was deeply searched and retrieved from protein data bank. Design of the protein and ligands structure were determined by the wizard panel of Glide [15] and 2D and 3D crystalline structure of ligands were obtained by screening in Maestro format. Optimizing of selected compounds at neutral pH, was strictly followed by the rules of LigPrep module of Schrodinger.
Glide
As a part of the mechanistic studies, affinities between protein to protein or with ligands were estimated by Glide, a tool to visualize, and to predict the interaction abilities. Structural and active sites of proteins were performed by using hierarchical sequence of filters to explore pose and characterization of selected receptors as defined on the grid [16].
Molecular docking
Protein-ligand docking is a technique of molecular modelling and docking was accomplished by using XP mode of Glide, a widely used realm and compressed of 5 taps. Scores and 3D structures were displayed in glide and energy evaluation was computed by calculating obtained Glide score. Docking was checked and scores were recorded. To determine free energy, docking score and conspicuous interactions with ligand was taken into accounts [17].
Free binding energy determination
To find free binding energy in order to verify the potential of biological actions of lead docked complexes, Maestro (MM-GBSA) was used. Individual molecules have been selected for the further examinations [18].
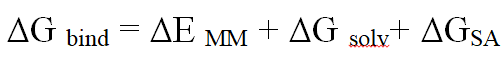
DFT analysis and ADME properties determination
DFT analysis was executed using Jaguar software. It covers MESP, HOMO and LUMO computation [19] and to analyze profiles of adsorption, distribution, metabolism, excretion (ADME) and response to toxicity were examined. Prior to operation, hit compounds in quick Prop mode, they were completely neutralized. Finally, QPPMDCK, QPLOG and QPPCaco measures were examined within the limit of Lipinski's rule five [20].
Molecular dynamic simulation
Based on the docking values, a high score complex was selected for the simulation process. To ascertain the stability of the complex, it was subjected to RMSD analysis [21]. RMSF scores for selected lead hit compounds; affinity nature was evaluated at 20 ns time modulation. On the ground of interacting power with ABL kinase binding sites were chosen for simulation. Using GROMACS 4.6.1 force field, behavior of the identified complex was tested [22].
Results and Discussion
Virtual screening
A series of screening was executed to recognize the strong inhibitor against ABL kinase protein using mechanistic screening workflow in Schrodinger. After filtering violated compounds, all the drug-like molecules were well docked with active sites of ABL kinase proteins. Results of screened drug-like compounds derived from 20 medicinal plants were listed in Table 1. The best docking score was found to be -11.959 kcal/mol and showed a good affinity with MET-318. Interaction poses were shown in Figure 1.

Figure 1: The docking results of 2D interaction diagram of the selected lead compound in the active site of ABL kinase protein.
| Compound ID | XP docking score (kcal/mol) | Glide score (kcal/mol) | Interacting residues | MM-GBSA ΔG Bind (kcal/mol) |
|---|---|---|---|---|
| 5281601 Apigenin Dimethylether |
-11.959 | -11.959 | MET-318 | -50.10 kcal/mol |
Table 1. Docking Score, Glide docking energy, interacting residues and Binding free energy score of Apigenin Dimethylether.
Binding free energy determination
Results showed that, a value of -50.10 kcal/mol was recorded as the binding free energy of ABL kinase – ligand complex. Table 1 also depicts obtained docking value and binding energy for the authenticity as an inhibitor of ABL kinase protein.
DFT computation
After optimizing the lead complex in PBP salvation environment, nucleophilic and electrophilic interaction sites for screening were performed. The obtained values of HOMO and LUMO were found to be 0.22 eV and -0.06 eV. To distinguish the interaction properties, mapping of stereo electronic support of ligand was encaged. As a result of selected compound MESP efficacy of Apigenin dimethyl ether, was found to be-14.80 kcal/mol. Table 2 indicates the value of HOMO, LUMO and MESP. Figure 2 displayed a diverse pose of the compound.

Figure 2. HOMO, LUMO and MESP parameter of Lead compound from DFT calculation.
| Compound ID | HOMO (eV) | LUMO (eV) | Solvation energy (kcal/mol) | MESP (kcal/mol) | |
|---|---|---|---|---|---|
| Most positive potential | Most negative potential | ||||
| 5281601 | -0.22 | -0.06 | -14.80 | -0.0176 | 0.0176 |
Table 2. HOMO, LUMO and MESP parameter of Lead compound from DFT calculation.
ADMET determination
For the successful marketing in the pharmaceutical field, ADME properties were approached and results were presented in Table 3. Findings indicating Apigenin dimethyl ether was suited for drug manufacturing and obeyed the limit of Lipinski rule 5, based on the molecular weight, consumption efficacy and hijacking ability across BBB of the drugs (Figure 3).

Figure 3. (a) Plots of Backbone RMSD of the selected ligand in the active site of ABL Kinase protein. (b) RMSF graph of selected protein ligand compound.
| Compound ID | Molecular Weight | Donor HB | Accept HB | QPlog Po/w | QPlogS | QPP Caco | Percent human oral absorption | Rule Of Five |
|---|---|---|---|---|---|---|---|---|
| 5281601 | 298.29 | 0.000 | 3.750 | 3.162 | -4.021 | 1272.2 | 100.00 | 0 |
Table 3. ADME analysis of top lead compound using QikProp.
Molecular dynamic simulation
It reveals that the identified complex was modulated around 10 ns interval, and then reached stableness in 15 ns at the final stage of the simulation. It is demonstrated that the screened compound is highly stable. RMSF analysis helped to calculate average fluctuation of the macromolecules, at residue level in the simulation process, Peaks eluted in RMSF plot denotes, residue of the protein that had been modulated often. As a result of RMSF, it obviously indicates, the majority of the residues of the compound modulated in less than 3 Angstrom units.
Conclusion
The current in silico approach has intricated a series of sequential screening and found lead hits compounds derived from 20 natural medicinal plants. Among them Andrographis paniculata was found to be more effective in arresting or attenuating the actions of ABL- kinase protein. The outcomes of the study can be considered as an alternate option for the existing therapy for cervical cancer. However, the active nature of bioactive compounds further validates their interactive ability in clinical prognosis.
Conflict of Interest
The authors declared that there are no conflicts of interest.
Acknowledgements
Acknowledge the financial support of DST-FIST [SR/ FST/LSI-667/2016(C)], DST-PURSE [SR/PURSE Phase 2/38 (G)] and MHRD-RUSA-phase 2.0 grant sanctioned vide [F.24-51/2014-U policy (TNMulti – Gen), Dept of Edn. Govt of India Dt, 09.10.2018] for the infrastructure facilities available in the Department of Bioinformatics, Alagappa University.
References
- Andrae B, Therese ML Andersson, Paul CL, Levent K, Lena S, Björn S, Walter Ryd, Joakim D, Sven T, and Pär S. Screening and Cervical Cancer Cure: Population Based Cohort Study. BMJ 2012; 344: 4-6.
- Barrón EV, Edgar RB, Ana LSS, Ana ME, Mariano GE, Ingrid M, Eligia J, Ana A, Miriam B, Rubén Z, Carlos GR, Juan CG, Susana KE, Martha PA, Jaime B. CDKN3 MRNA as a Biomarker for Survival and Therapeutic Target in Cervical Cancer. PLoS ONE 2015; 10: 1-26.
- Caterina R, Guillermo DC, Antonio HD, Sofia ML, Katja EL, Päivi T, Merja P, Vesa H, Johannes V, Heiko R, Christian DM, Manuel N, José JF, María LS. Marine Microalgae: Promising Source for New Bioactive Compounds. Mar Drugs 2018; 16: 1-12.
- Deshpande A, Keerthi K, Jayanthi S. Insilico Docking Studies for Investigating the Anticancer Properties of Plant Based Compounds against Cervical Cancer. Research J Pharm Biol Chem Sci 2017; 8: 1161-1166.
- Goff SP, Eli G, Owen NW, David B. Structure of the Abelson Murine Leukemia Virus Genome and the Homologous Cellular Gene: Studies with Cloned Viral DNA. Cell 1980; 22: 777-785.
- Greenwell M, Rahman PKS. Medicinal Plants : Their Use in Anticancer Treatment. Int J Pharm Sci Res 2015; 6: 4103-4112.
- Gschwend DA, Andrew CG, Irwin DK. Molecular Docking towards Drug Discovery. J Mol Recog 1996; 9: 175-186.
- Lin J, Arlinghaus R. Activated C-Abl Tyrosine Kinase in Malignant Solid Tumors. Oncogene 2008; 27: 4385-4391.
- Muhammed Bah Yahya. Drug Abuse among Street Children. J Clin Res HIV AIDS Prev 2018; 3: 10-19.
- Pendergast Ann Marie. The Abl Family Kinases: Mechanisms of Regulation and Signaling. Adv Cancer Res 2002; 85: 51-100.
- Simpson L, Xiang H, Michael P, Xiaoke H, Steven CC, Ximing JY, Elizabeth JP, Raymond CB.Renal Medullary Carcinoma and ABL Gene Amplification. J Urol 2005; 173: 1883-1888.
- Srinivasan D, Rina P. Activation of Abl Tyrosine Kinases Promotes Invasion of Aggressive Breast Cancer Cells. Cancer Res 200; 66: 5648-5655.
- Yamada T, Manos MM, Peto J, Greer CE, Munoz N, Bosch FX, Wheeler CM. Human Papillomavirus Type 16 Sequence Variation in Cervical Cancers: A Worldwide Perspective. J Virol 1997; 71: 2463-2472.
- Kumar, Avinash, EktaRathi, Suvarna GK. E-Pharmacophore Modelling, Virtual Screening, Molecular Dynamics Simulations and in-Silico ADME Analysis for Identification of Potential E6 Inhibitors against Cervical Cancer. J Mol Stru 2019; 1189: 299-306.
- Madhavi SG, Matvey A, Tyler D, Ramakrishna A, Woody S. Protein and Ligand Preparation: Parameters, Protocols, and Influence on Virtual Screening Enrichments. J Comput Aided Mol Des2013; 27: 221-234.
- Schulz-Gasch Tanja, Martin Stahl. Binding Site Characteristics in Structure-Based Virtual Screening: Evaluation of Current Docking Tools. J Mol Model 2013; 9: 47-57.
- Paul DL, Michelle LL, Jamal C. Saeh. Accurate Prediction of the Relative Potencies of Members of a Series of Kinase Inhibitors Using Molecular Docking and MM-GBSA Scoring. J Med Chem 2016; 49: 4805-4808.
- Kumar N, Goel N, Yadav TC, Pruthi V. Quantum Chemical, ADMET and Molecular Docking studies of ferulic acid amide derivatives with a novel anti-cancer drug target. Med Chem Res 2017; 26: 1822-1834.
- Nisha CM, Kumar A, Vimal A, Bai BM, Pal D, Kumar A. Docking and ADMET prediction of few GSK-3 inhibitors divulges 6-bromoindirubin-3-oxime as a potential inhibitor. J Mol Graph Model 2016; 65: 100-107.
- Kumar N, Goel N, Yadav TC, Pruthi V. Quantum Chemical, ADMET and Molecular Docking studies of ferulic acid amide derivatives with a novel anti-cancer drug target. Med Chem Res 2017; 26: 1822-1834.
- Amala M, Sundaraj R, Dhamodharan P, Kanagarajan S, Jeyaraman J. Identification of Anti-Filarial Leads against Aspartate Semialdehyde Dehydrogenase of WolbachiaEndosymbiont of BrugiaMalayi: Combined Molecular Docking and Molecular Dynamics Approaches. J Biomol Struc Dyn 2019; 37: 394-410.
- Jayaraj, John M, Gopinath K, Jung Kul Lee, Karthikeyan M. In Silico Identification and Screening of CYP24A1 Inhibitors: 3D QSAR Pharmacophore Mapping and Molecular Dynamics Analysis. J Biomol Struc Dyn 2019; 37: 1700-1714.