| There are several signaling pathways identified to play a role in the self-renewal abilities of breast cancer stem cells. The Hedgehog (Hh)- , Notch- , Wingless-type (Wnt)/ß- catenin- and the inflammatory pathways are examples of these signaling pathways (Figure 2) [8,16-19]. |
Notch Signaling Pathway
|
| The Notch signaling pathway plays a critical role in stem cell fate determination, cell cycle progression and normal embryonic development [17,19]. Notch pathway action has been indicated in bCSC models where mammary tumor formation is promoted due to the change in morphogenetic properties caused by Notch-4 overexpression [8,20]. The Notch pathway also contributes to the maintenance of breast cancer stem cells and other tumor stem cells by interacting with erythropoietin and the Erb2 (HER2) promoter binding sequence [20,21]. |
| There are four transmembrane Notch receptors (Notch1- Notch4) and 5 ligands consisting of Delta-like proteins (DLL1, DLL3, DLL4) and Jagged-proteins (JAG1, JAG2) [17-19]. The ligands bind to the outer membrane receptor proteins resulting in the intramembrane cleavage of the receptor. The latter is due to a disintegrin and metalloproteinase (ADAM)/?-secratase proteolytic cleavages [17,21]. This allows translocation of the intracellular domain (NCID) toward the nucleus [17- 19]. In the nucleus, NCID interacts with mastermind-like proteins 1/2 and 3 (MAML1/2/3), as well as CSL (CBF1/ Lag1/RBP-J?) factors. This leads to the transcriptional activation of Notch genes, including cyclin D or nuclear factor kappa-light-chain-enhancer of activated B cells (NF-?B) (Figure 2A) [17]. Cyclin D regulates cell cycle progression and overexpression due to increased Notch pathway activation demonstrating uncontrolled growth [19]. |
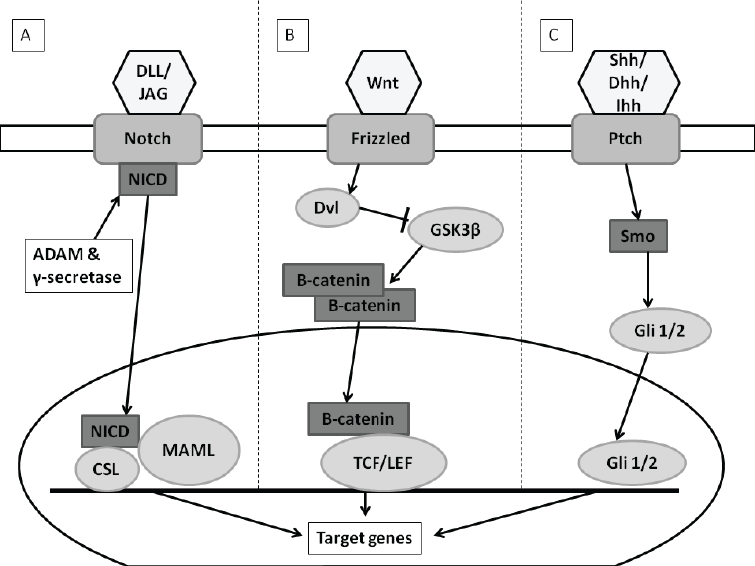 |
| Figure 2: Notch, Wnt and Hedgehog signaling pathways. A) Notch signaling pathway: Notch ligands (DLL/JAG) bind to Notch receptors allowing for NCID to be released into the cytoplasm from membrane. NCID is translocated to nucleus where it interacts with MAML/CSL factors. Transcription of Notch genes is activated. B) Wnt signaling pathway (in presence of Wnt ligand): Wnt ligand binds to and activates the Axin/Frizzled/LRP complex which allows for Dvl to be released. Dvl inhibits phosphorylation of ß-catenin. ß-catenin accumulates in cytoplasm and is translocated to the nucleus where it binds to TCF/LEF. Transcriptions of Wnt genes are activated. C) Hedgehog signaling pathway (In presence of Hh ligand): Hh ligand relieves inhibited Smo through binding to Ptch which allows Gli 1/2 transcription factor to be translocated to the nucleus and activates Hh gene transcription. Image created with Microsoft PowerPoint® 2010 (Microsoft Corporation, California, United States of America) |
Wnt/ß-catenin Signaling Pathway
|
| Alexander et al. (2004) demonstrated that up regulation of the Wnt/ß-catenin pathway contributes to breast cancer by playing a role in breast cancer stem cell self-renewal in transgenic mice [22]. They observed an increase in the number of mammary stem cells due to activated ß-catenin in mammary epithelium or overexpression of Wnt ligands in mammary stroma [22]. |
| In bCSC amplified amounts of ß-catenin are present along with increased Wnt ligand expression [22]. In the Wnt/ß-catenin pathway (canonical pathway), Wnt ligand binds to a receptor complex of Frizzled receptors and low-density lipoprotein receptor-related proteins 5 and -6 (LRP5/6) [17,19,21]. In the absence of the Wnt ligand a destruction complex forms resulting in ß-catenin accumulating in the cytoplasm that cannot be translocated to the nucleus [17,19]. ß-Catenin then binds to glycogen synthase kinase (GSK-3ß), axin and adenomatous polyposis coli (APC) which form the ß-catenin destruction complex [19]. |
| ß-Catenin is phosphorylated through GSK-3ß which leads to degradation of the complex [21]. Presence of the Wnt ligand activates dishevelled (Dvl) inhibits the destruction complex resulting in ß-Catenin translocation to the nucleus (Figure 2B). In the nucleus ß-catenin binds and activates the transcription factor T cell factor/lymphoid enhancing factor (TCF/LEF) resulting in the transcription of the Wnt genes such as cyclin D and fibronectin [18,21]. |
Hedgehog Signaling Pathway
|
| The Hedgehog pathway (Hh) was first discovered in Drosophila as being essential in segmental embryo patterning [19,21]. Since then it has been discovered that the Hh pathway also plays a role in cell proliferation, migration and differentiation [17]. In several solid tumor CSC models including breast, increased activation of the Hh pathway has been identified [8]. The 3 ligands of the Hh pathway are the Sonic (SHH), the Indian (IHH) and Desert (DHH). Other components are the Patched membrane receptor (PTCH1/2) and the Smoothened signal transducer (Smo) which form a complex with each other in the absence of the Hh ligands [17,19]. PTCH will inhibit Smo which prevents the modulation of the gliomaassociated oncogene family zinc finger 1/2/3 (GLI 1/2/3) transcriptions factors [17,19]. GLI subsequently form a multi-protein complex with Fused (FU), Suppressor of Fused (SUFU) and Drosophila costal 2 (Cos 2). When the Hh ligands are present, Smo will not be inhibited [17, 19]. This allows GLI to translocate to the nucleus (Figure 2C) where it can activate many genes for transcription including cyclin D and cyclin E [19]. |
Inflammatory Signaling Pathway
|
| Chemokine?and/or cytokine-mediated inflammatory signaling pathways play a role in breast cancer stem cell maintenance [16]. Some inflammatory genes that are involved in this regulation include NF-?B, Interleukin 6 and 8 (IL6/8) and tumor-necrosis factor a (TNF-a) [16]. IL6 plays a role in the self-renewal of breast cancer stem cells through a paracrine/autocrine Notch-3/JAG-1 loop [16]. IL6 forms a positive feedback loop with NF-?B, maintaining mammosphere formation as demonstrated by Iliopoulos et al. (2009) [23]. High IL6 levels are maintained by means of IL6 transcription activation by NF-?B. Reciprocally, high levels of IL6 activate NF-?B [23]. TNF-a and interferon-? (INF-?) pathways are up regulated in bCSC and the two pathways activate NF-?B and vice versa [24]. A pro-inflammatory chemokine, IL8, binds to the CXCR1 receptor activating protein kinase B (Akt) resulting in ß-catenin translocation to the nucleus forming a complex with T-cell factor (TCF) [16]. |
Chemoresistance
|
| Conventional chemotherapies used to treat aggressive breast cancers may be effective initially, but, over time many patients will relapse [25]. Although most of the breast cancer cells are killed by cytotoxic agents, bCSC survive. These bCSC then have the ability of regrowth, forming new tumors and causing patient relapses [25]. Chemoresistance of bCSC can be divided into two main groups namely intrinsic resistance due to genetic alterations and extrinsic resistance including microenvironment influences [26]. Intrinsic resistance include overexpression of adenosine triphosphate (ATP) binding cassette transporter proteins (ABC transporter), the adapted deoxynucleotide acid (DNA) repair mechanism, an altered cell cycle, overexpression of aldehyde dehydrogenase 1 (ALDH1) and resistance to apoptosis [26-28]. The extrinsic group includes all microenvironment influences such as hypoxia or epithelial-mesenchymal transitions (EMT) that lead to chemoresistance [27]. |
Intrinsic Resistance
|
| The small population of cells that survive chemotherapy treatment is potentially due to ABC transporters found on bCSC [26]._ENREF_30 ABC transporter proteins use energy gained from ATP binding and hydrolysis to transport substrates such as anticancer drugs [27]. ABC transporters rapidly cause the efflux of these chemotherapeutic drugs out of the bCSC leading to increased chemoresistance [29]. Hirschmann-Jax et al. (2004) reported high levels of ABCG2 in bCSC accompanied with an improved survival rate due to increased capacity to expel cytotoxic drugs and the ability to confer cellular resistance to antineoplastic drugs [30]. High levels of P-glycoprotein (Pgp) also confer resistance to chemotherapeutic drugs by influencing many cellular processes like the p53 network which mediates chemoresistance [26]. Thus, the bCSC that express high levels of ABC transporters (ABCG2, Pgp) survive chemotherapy [26]. New tumors with a chemoresistant phenotype regrow due to cells that survived and mutations caused by chemotherapy [31]. Through use of an efflux pump mechanism, ABC transporter molecules protect the bCSC against any damage caused by chemotherapeutic drugs [28,31]._ENREF_30 |
| ALDH, as previously mentioned, is a biomarker for breast cancer stem cell identification [28]. Through use of aldefluor assays Ginestier et al. (2007) indicated that breast cancer cells that were highly tumorigenic were ALDH+. These bCSC had the same properties of self-renewal and differentiation when compared to CSC [32]. This detoxifying enzyme oxidizes aldehydes to form carboxylic acids. ALDH1A1 and ALDH3A1 play a critical role in the self-protection and differentiation of stem cells through the conversion of retinol to retinoic acid [33]. ALDH1 has the ability of metabolizing chemotherapeutic agents, especially cyclophosphamide through the conversion of aldophosphamide to carboxyphosphamide and thereby eliminating the toxic effects of the metabolites acrolein and phosphoramide mustard [28, 33, 34]. Metastasis is associated with overexpression of ALDH accompanied by a poor prognosis [35]. ALDH over expression has been indicated as one of the causes of chemoresistance [28,33]. |
| Altered cell cycle kinetics is another intrinsic resistance mechanism found in bCSC [27,36]. This allows bCSC to escape death from chemotherapeutic agents that target rapidly dividing cells such as normal breast cancer cells [27,28,36]. This dormant state of bCSC can also explain the relapses of breast cancer after long-periods of time [36]. A proficient DNA repair mechanism in CSC is another intrinsic resistance mechanism [37]. bCSC use increased checkpoint (ChK) activation of ChK1 and ChK2 allowing escape from mitotic catastrophe after chemotherapy treatment and to repair their DNA proficiently [27,37,38]. This state of dormancy and proficient DNA repair mechanism can contribute to the chemoresistance of bCSC [36]. |
Extrinsic Resistance
|
| The indirect mechanism of chemoresistance (extrinsic) takes into account the microenvironment and its influence on bCSC [26,27]. The interaction between the microenvironment and cancer stem cells is a dynamic process leading to continuous remodeling of both [27]. Epithelial-mesenchymal transitions (EMT) play a critical role in bCSC chemoresistance and development of cancer metastasis [27]. Paracrine-acting signals such as self-renewal pathways (Notch/Wnt/Hh) induce EMT by activation of a transcriptional complex resulting in a cytoskeleton rearrangement towards a mesenchymal-like phenotype [39-41]. Cells typically found in the tumorstroma that undergo these morphological changes, will gain pro-metastatic characteristics increasing stem-cell like markers and clonogenicity [27, 42]. |
| Hypoxia has also been identified as a regulator of CSC since the tumor growth is faster when compared to blood supply resulting in a hypoxic environment [27,43]. Self-renewal properties of both stem cells and CSC are promoted by hypoxia. In the presence of low oxygen levels, hypoxiainducible factor (HIF) is activated resulting in new blood vessel formation (angiogenesis) and promotes a prosurvival phenotype [15]. New blood vessels limit drug perfusion, due to their abnormal architecture resulting in lower concentrations of chemotherapeutics drugs in tumors [27,43]. In addition, HIF-1 contributes to chemoresistance in bCSC through mechanisms of genomic instability and, abnormal cell cycles [15]. HIF is capable of reprogramming non-stem-like cells to have more stem cell-like traits such as self-renewal capabilities by inducing the expression of key stem cell genes like octamer-binding transcription factor 4 (Oct4) and myelocytomatosis cellular oncogene (c-Myc) [43]. In addition, hypoxia creates niches for CSC by means of increased lysyl oxidase (LOX) production [26,27,43]. Thus indirectly the microenvironment and hypoxia contribute to chemoresistance [27]. |
Applications
|
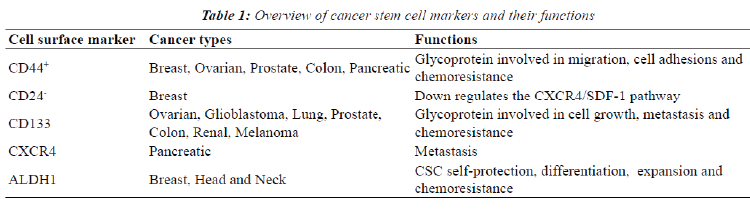
| For optimal incapacitation of chemoresistance in cancer, the sub-population of CSC should be eliminated [21]. The identification of CSC is therefore required since both stem cells and CSC have similar signaling pathways and mechanism [37]. A variety of cell surface markers are specific for bCSC and these are currently being used for identification (Table 1) including clusters of differentiation 44, 24 and 133 [8]. Highly tumorigenic breast cancer types such as those with a BRCA1 defect have been found to express high levels of CD44 and no or low amounts of CD24 (CD44+/CD24-) [8]. _ENREF_15 CD44 is a cell-surface glycoprotein involved in migration and cell adhesions and in addition binding of CD44 to hyaluronic acid (HA) is essential in tumor progression by inhibiting apoptosis [21,44]. CD24, a glycoprotein, down regulates the CXCR4/SDF-1 pathway which contributes to breast metastasis due to its role in cell migration [45]. Processes that are essential to tumor metastasis such as their chemoattraction, adhesion and locomotion of malignant cells are regulated by the pleiotropic effects exerted by SDF-1 [46]. ALDH1 is also highly expressed in bCSC, specifically in estrogen receptor negative breast cancers and correlates with a lower overall survival rate [47]. This isoenzyme is not only used to identify CSC but also plays a role in and CSC self-protection, differentiation and expansion [21,48]. Epithelial-specific antigen (ESA), CD133 (prominin-1) and CXCR4 are other biomarkers used to identify breast cancer stem cells [8,14]. ESA is used to differentiate between benign reactive epithelial cells and epithelial cancer cells. Populations containing ESA+/ CD44high/CD24low biomarkers have an enhanced capacity for tumor and mammosphere formation [8,14]. CD133+ cells containing CSC characteristics have been identified in triple negative breast cancers and also show increase survival in vitro [8,21]. |
| By combining conventional and CSC targeted therapies, it will improve efficacy of cancer therapy (Figure 3) [5]. These future combination cancer therapies may help improve cancer prognosis, specifically for metastatic cancers [5, 49]. They may potentially reduce the chemoresistance of cancer and thereby also improve overall survival with a decrease in relapses [5,37,49]. |
Controversies
|
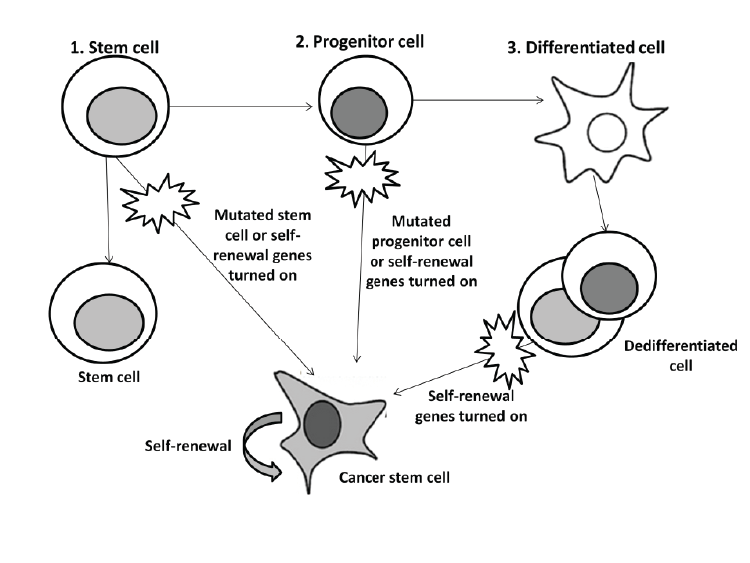
| The existence of CSC has been debated since 1970 when scientists discovered two sub-populations in leukemia cells [50]. One school of thought endorses the existence of CSC and that they are a crucial target for cancer therapy. Opponents to this theory question the separate entirety of CSC and argue their importance in cancer pathology and treatment [51]. One drawback is that experiments for CSC have only been conducted on immunodeficient mice raising concerns since results do not represent the reality of cancer initiation and progression in humans [49]. Other reports demonstrated that CSC does not necessarily lead to tumor formation, but rather that the clonal evolution model (stochastic) is a better explanation for this subpopulation of cells origin [50,52]. This model suggests that the heterogeneity found in cancer cells is due to variation in the levels of transcription factors that vary amongst the cells [50]. The CSC hypothesis still remains an attractive model despite all these controversies. Thus, continuing research is still required for distinguishing between stem cells and CSC for differential drug activity [8,37]. Should this population of cells be fully defined and characterized, they could provide a potential target for eradicating cancer. |
 |
Conclusion
|
| Breast cancer recurrence occurs 40% of the time in part due to resistance to chemo- and radiation therapies [53]. By better understanding the CSC phenotype causing chemoresistance, more improved cancer treatments can be developed to prevent relapses [9, 14]. One method to overcome chemoresistance in bCSC is for future cancer therapies to focus on drugs that inhibit signaling pathways (Wnt/Hh/Notch) responsible for the self-renewal of CSC [37, 49]. The role that the microenvironment and hypoxia play should also be taken into account due to their contribution to chemoresistance [37]. |
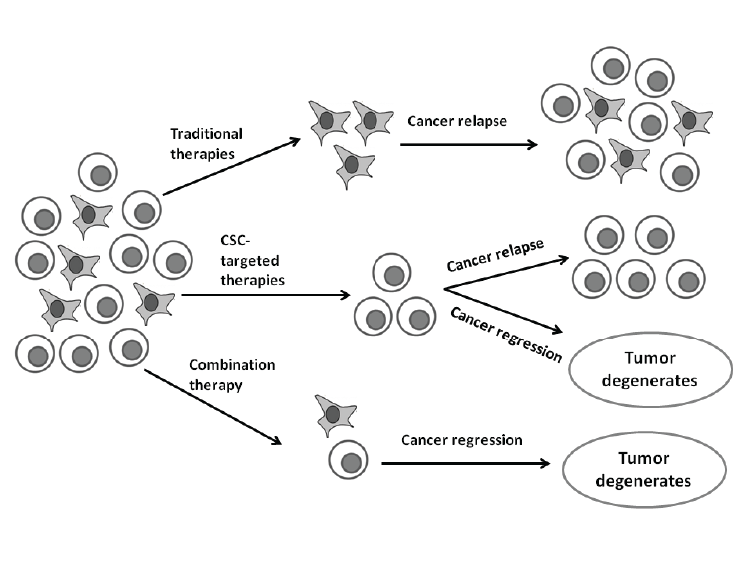 |
| Figure 3: Therapeutic implications of combination therapies. Traditional therapies fail to target CSC and mostly only kill differentiated cancer cells. CSC can thus survive and could lead to relapses. CSC-targeted therapies either eliminate CSC or result in differentiation where after these differentiated cancer cells may also undergo cell death via apoptosis. However, combination therapy could be most effective for tumor elimination. Image created with Microsoft PowerPoint® 2010 (Microsoft Corporation, California, United States of America) |
| By better understanding the CSC phenotype causing chemoresistance, more improved cancer treatments can be developed to prevent relapses [9,14]. One method to overcome chemoresistance in bCSC is for future cancer therapies to focus on drugs that inhibit signaling pathways (Wnt/Hh/Notch) responsible for the self-renewal of CSC [37,49]. The role that the microenvironment and hypoxia play should also be taken into account due to their contribution to chemoresistance [37]. |
Acknowledgement
|
| This work was supported by grants from the Medical Research Council of South Africa, the Cancer Association of South Africa, National Research Foundation and the Struwig-Germeshuysen Cancer Research trust of South Africa. |
|
|
References
- 1. National Cancer Institute. SEER Cancer Statistics Factsheets: Breast Cancer.
- 2. Yerushalmi R, Hayes MM, Gelmon KA. Breast carcinomarare types: review of the literature. Ann Oncol 2009; 20: 1763-1770.
- 3. Parise CA, Caggiano V. Breast Cancer Survival Defined by the ER/PR/HER2 Subtypes and a Surrogate Classification according to Tumor Grade and Immunohistochemical Biomarkers. J Cancer Epidemiol 2014; 2014:1-11.
- 4. Basu S, Chen W, Tchou J, Mavi A, Cermik T, Czerniecki B, Schnall M, Alavi A. Comparison of triple-negative and estrogen receptor-positive/progesterone receptor-positive/ HER2-negative breast carcinoma using quantitative fluorine-18 fluorodeoxyglucose/positron emission tomography imaging parameters. Cancer Cell Inter 2008; 112: 995-1000.
- 5. Han L, Shi S, Gong T, Zhang Z, Sun X. Cancer stem cells: therapeutic implications and perspectives in cancer therapy. Acta Pharmaceutica Sinica B 2013; 3: 65-75.
- 6. Takahashi R, Takeshita F, Fujiwara T, Ono M, Ochiya T. Cancer stem cells in breast cancer. Cancers 2011; 3: 1311- 1328.
- 7. Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CHM, Jones DL, Visvader J, Weissman IL, Wahl GM. Cancer Stem Cells-Perspectives on Current Status and Future Directions: AACR Workshop on Cancer Stem Cells. Cancer Res 2006; 66: 9339-9344.
- 8. Iqbal J, Chong PY, Tan PH. Breast cancer stem cells: an update. J Clin Pathol 2013; 66: 485-490.
- 9. National Institutes of Health. Are stem cells involved in cancer?
- 10. Allan AL, Vantyghem SA, Tuck AB. Tumor dormancy and cancer stem cells: implications for the biology and treatment of breast cancer metastasis. Breast Dis 2007; 26: 87-98.
- 11. Li F, Tiede B, Massagué J, Kang Y. Beyond tumorigenesis: cancer stem cells in metastasis. Cell Res 2007; 17: 3-14.
- 12. Yu J, Vodyanik MA, Otto KS, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, Slukvin II, Thomson JA. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007; 318: 1917-1920.
- 13. Hermann PC, Huber SL, Herrler T, Herrier T, Aicher A, Ellwart JW, Guba M, Bruns CJ, Heeschen C. Distinct Populations of Cancer Stem Cells Determine Tumor Growth and Metastatic Activity in Human Pancreatic Cancer. Cell stem cell 2007; 1: 313-321.
- 14. Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci 2003; 100: 3983-3988.
- 15. Crowder SW, Balikov DA, Hwang YS, Sung HJ. Cancer Stem Cells Under Hypoxia as a Chemoresistance Factor in the Breast and Brain. Curr Pathobiol Rep 2014; 2: 33-40.
- 16. Hinohara K, Gotoh N. Inflammatory signaling pathways in self-renewing breast cancer stem cells. Curr Opin Pharmacol 2010; 10: 650-654.
- 17. Karamboulas C, Ailles L. Developmental signaling pathways in cancer stem cells of solid tumors. Biochim Biophys Acta 2013; 1830: 2481-2495.
- 18. Takebe N, Ivy SP. Controversies in Cancer Stem Cells: Targeting Embryonic Signaling Pathways. Clin Cancer Res 2010; 16: 3106-3112.
- 19. Liu S, Dontu G, Wicha MS. Mammary stem cells, selfrenewal pathways, and carcinogenesis. Breast Cancer Res 2005; 7: 1-10.
- 20. Guo S, Liu M, Gonzalez-Perez RR. Role of Notch and its oncogenic signaling crosstalk in breast cancer. Biochimica et Biophysica Acta 2011; 1815: 197-213.
- 21. Hu Y, Fu L. Targeting cancer stem cells: a new therapy to cure cancer patients. Am J Cancer Res 2012; 2: 340-356.
- 22. Liu BY, McDermott SP, Khwaja SS, Alexander CM. The transforming activity of Wnt effectors correlates with their ability to induce the accumulation of mammary progenitor cells. Natl Acad Sci USA 2004; 101: 4158-4163.
- 23. Iliopoulos D, Hirsch HA, Struhl K. An epigenetic switch involving NF-kappaB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell Res 2009; 139: 693-706.
- 24. Murohashi M, Hinohara K, Kuroda M, Isagawa T, Tsuji S, Kobayashi S, Umezawa K, Tojo A, Aburatani H, Gotoh N. Gene set enrichment analysis provides insight into novel signalling pathways in breast cancer stem cells. Br J Cancer 2010; 102: 206-212.
- 25. Li X, Lewis MT, Huang J, Gutierrez C, Osborne CK, M Wu, Hilsenbeck SG, Pavlick A, Zhang X, Chamness GC, Wong H, Rosen J, Chang JC. Intrinsic Resistance of Tumorigenic Breast Cancer Cells to Chemotherapy. J Natl Cancer Inst 2008; 100: 672-679.
- 26. Rebucci M, Michiels C. Molecular aspects of cancer cell resistance to chemotherapy. Biochem Pharmacol 2013; 85: 1219-1226.
- 27. Maugeri-Saccà M, Vigneri P, De Maria R. Cancer Stem Cells and Chemosensitivity. Clin Cancer Res 2011; 17.
- 28. Economopoulou P, Kaklamani VG, Siziopikou K. The Role of Cancer Stem Cells in Breast Cancer Initiation and Progression: Potential Cancer Stem Cell-Directed Therapies. Oncol 2012; 17: 1394-1401.
- 29. Dean M. ABC Transporters, Drug Resistance, and Cancer Stem Cells. J Mammary Gland Biol Neoplasia 2009; 14: 3-9. 30. Hirschmann-Jax C, Foster AE, Wulf GG, Nuchtern JG, Jax TW, Gobel U, Goodell MA. A distinct ??side population?? of cells with high drug efflux capacity in human tumor cells. Proc Natl Acad Sci USA 2004; 101: 14228-14233.
- 31. Chuthapisith S, Eremin J, El-Sheemey M, Eremin O. Breast cancer chemoresistance: Emerging importance of cancer stem cells. Surg Oncol 2010; 19: 27-32.
- 32. Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, Jacquemier J, Viens P, Kleer CG, Liu S, Schott A, Hayes D, Birnbaum D, Wicha MS, Dontu G. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007; 1: 555-567.
- 33. Croker AK, Allan AL. Inhibition of aldehyde dehydrogenase (ALDH) activity reduces chemotherapy and radiation resistance of stem-like ALDHhiCD44+ human breast cancer cells. Breast Cancer Res Treat 2012; 133.
- 34. Agarwal DP, Eitzen UV, Meier-Tackmann D, Goedde HW. Metabolism of Cyclophosphamide by Aldehyde Dehydrogenases. Enzymology and Molecular Biology of Carbonyl Metabolism 5. Advances in Experimental Medicine and Biology. 1995; 372: 115-122.
- 35. Charafe-Jauffret E, Ginestier C, Iovino F, Tarpin C, Diebel M, Esterni B, Houvenaeghel G, Extra JM, Bertucci F, Jacquemier J, Xerri L, Dontu G, Stassi G, Xiao Y, Barsky SH, Birnbaum D, Viens P, Wicha MS. Aldehyde dehydrogenase 1-positive cancer stem cells mediate metastasis and poor clinical outcome in inflammatory breast cancer. Clin Cancer Res 2010; 16: 45-55.
- 36. Clevers H. The cancer stem cell: premises, promises and challenges. Nature Med 2011; 17: 313-319.
- 37. Maugeri-Saccà M, Zeuner A, De Maria R. Cancer Stem Cells from Solid Tumors: New Tools to Fight Cancer. AACR 2011: 133-138.
- 38. Eyler CE, Rich JN. Survival of the Fittest: Cancer Stem Cells in Therapeutic Resistance and Angiogenesis. J Clin Oncol 2008; 26: 2839-2845.
- 39. Ohta H, Aoyagi K, Fukaya M, Danjoh I, Ohta AA, Isohata N. Cross talk between hedgehog and epithelial-mesenchymal transition path-ways in gastric pit cells and in diffuse-type gastric cancers. Br J Cancer 2009; 100: 389-398.
- 40. Wang Z, Li Y, Kong D, Banerjee S, Ahmad A, Azmi AS. Acquisition of epithelial-mesenchymal transition phenotype of gem-citabine-resistant pancreatic cancer cells is linked with activation of the notch signaling pathway. Cancer Res 2009; 69.
- 41. Gupta S, Iljin K, Sara H, Mpindi JP, Mirtti T, Vainio P. FZD4 as a mediator of ERG oncogene-induced WNT signaling and epithelial-to-mesenchymal transition in human prostate cancer cells. Cancer Res 2010; 70: 6735-6745.
- 42. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, AY AY Zhou. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell Cycle 2008; 133: 704-715.
- 43. Heddleston JM, Li Z, McLendon RE, Hjelmeland AB, Rich JN. The hypoxic microenvironment maintains glioblastoma stem cells and promotes reprogramming towards a cancer stem cell phenotype. Cell Cycle 2009; 8: 3274-3284.
- 44. Orian-Rousseau V. CD44, a therapeutic target for metastasising tumours. Euro J Cancer 2010; 46: 1271-1277.
- 45. Hermann PC, Bhaskar S, Cioffi M, Heeschen C. Cancer stem cells in solid tumors. Sem Cancer Biol 2010; 20: 77-84.
- 46. Kucia M, Jankowski K, Reca R, Wysoczynski M, Bandura L, Allendorf DJ, Zhang J, Ratajczak J, Ratajczak MZ. CXCR4? SDF-1 signalling, locomotion, chemotaxis and adhesion. J Mol Histo 2004; 35: 233-245.
- 47. Nguyen NP, Almeida FS, Chi A, Nguyen LM, Cohena D, Karlsson U, Vinh-Hung V. Molecular biology of breast cancer stem cells: Potential clinical applications. Cancer Treat Rev 2010; 36: 485-491.
- 48. Ma I, Allan AL. The Role of Human Aldehyde Dehydrogenase in Normal and Cancer Stem Cells. Stem Cell Rev Rep 2011; 7: 292-306.
- 49. Charafe-Jauffret E, Monville F, Ginestier C, Dontu G, Birnbaum D, Wicha MS. Cancer Stem Cells in Breast: Current Opinion and Future Challenges. Pathobiol 2008; 75: 75-84.
- 50. Rowan K. Are Cancer Stem Cells Real? After Four Decades, Debate Still Simmers. J Natl Cancer Inst 2009; 101: 546- 547.
- 51. CT Jordan. Cancer Stem Cells: Controversial or Just Misunderstood? Cell stem cell 2009; 4: 203-205.
- 52. Quintana E, Shackleton M, Sabel MS, Fullen DR, Johnson TM, Morrison SJ. Efficient tumor formation by single human melanoma cells. Nature Med 2008; 488: 522-526.
- 53. Smalley M, Piggott L, Clarkson R. Breast cancer stem cells: Obstacles to therapy. Cancer Lett 2013; 338: 57-62.
|
-
|