ISSN: 0970-938X (Print) | 0976-1683 (Electronic)
Biomedical Research
An International Journal of Medical Sciences
Research Article - Biomedical Research (2017) Volume 28, Issue 8
Aiming Human Bocavirus VP-2 protein via combination of pharmacophore, molecular docking and ADME-toxicity methodologies for the identification of optimized and potent drug candidate.
Maulana Azad National Institute of Technology, Bhopal, Madhya Pradesh, India
Accepted date: December 28, 2016
Human bocavirus (HBoV) belongs to a parvoviridae family. It has been detect primarily in children with acute lower respiratory tract infection, but its occurrence, clinical profile, and role as a causative agent of respiratory tract disease are not clear. Currently, there is no treatment or vaccination. The leading objectives of the study were to describe the Human bocavirus VP 2 protein structure and its properties and discover the possible small molecule that can be additional use as a possible new drug candidate for dealing with infection produced by Human bocavirus 1. Small molecules were used as ligands and molecular dockings were made with the assistance of docking tool i.e., AutoDock. The molecule presenting the finest binding energies (zinc64624173 -7.99, zinc4099018 -7.88 and zinc64624174 -7.87) were used a possible new drug candidate. These possible drug candidates used to produce a pharmacophore model for small molecule ligands with the support of LigandScout software. Then pharmacophore based virtual screening was done against the already formed ligand library from ZINC database, so we get the finest hits or leads for our pharmacophore molecules. These hits or leads has been use as ligands and then exposed to molecular docking. The finest molecule was ZINC72330347-10.29 (Binding Energy) was found, after getting the best hits ADMET was analysis. Those molecules can use as an inhibitor to Human bocavirus VP 2 protein.
Keywords
Human Bocavirus VP2 protein, Molecular docking, Pharmacophore, LigandScout, SiteHound-web, ADME-Tox analysis.
Introduction
In 2005, Allander et al. [1] described the discovery of a new virus in paediatric respiratory nasopharyngeal aspirates, which knows as human bocavirus (HBoV) and was shown to remain a member of the parvovirus family. Three additional species, titled HBoV2–4, recognized subsequently. Former studies have shown that HBoV1 is a causative agent of respiratory symptoms in children and adults. HBoV2 and similarly HBoV3 were related to gastroenteritis but HBoV2 was predominant [2]. At present, the most common technique for diagnosis of HBoV is PCR, which is only available in research laboratories. There are not any standardized or FDA-approved assays for HBoV and the finest primer set and methodology has not been conclusively established [3]. From literature it was found that virus protein 2 presented major immunoreactivity [4]. The mode of infection of human bocavirus remains unfamiliar resulting ambiguity. Human bocavirus affects the human respiratory tract and it replicates in epithelial cells. In animals, it is found in respiratory and gut epithelium and lymphoid tissues.
In this study, we have used small molecules as ligands. Small molecules deliver the ideal platform for iterative methods to the design of inhibitors of a particular bacterial target protein, through structure-based drug design [5]. Small molecules are essential molecules and are important in regulating many biological processes. The tools of molecular biology have dramatically improved the discovery and development of fresh biopharmaceuticals. The most observable difference between small-molecule drugs (SMDs) and biomolecular drugs is size, like the difference in weight between a bicycle and a business jet [6]. Secondary metabolites use as Small molecules like Alkaloids, Glycoside, Lipids, Non-ribosomal peptides, Phenazines, Natural phenols (including flavonoids), Polyketide, Terpenes, including steroids, Tetrapyrroles [6]. We cannot perform lackhs of molecules docking it will take time. Pharmacophore modelling can be used to find out similar molecule. Pharmacophore modelling is done with the help of LigandScout [7]. Pharmacophore models have shown to be an actual comprehensive and thus influential representation of small molecule binding [8]. The ligand-based method inherently involves the flexible alignment of molecules, which, on the single hand, can be completed simply by taking into account atom helps or through other methods that are dissimilar to pharmacophore representations [9]. Pharmacophore modeling and virtual screening is a low-cost and fast substitute powerful tool to recognize the potential lead for several targets. Therefore, in this study, ligand and structure-based pharmacophore models were generated and validated by the test and decoy sets in a wide range of applications. Virtual screening was established to be successful method especially when shared with molecular docking studies [10]. Docking used to predict the certain conformation and binding free energy of small molecules toward the target. Single docking experiments remain useful for exploring the function of the target, and virtual screening, in which a huge library of compounds are docked and ranked, may remain used to identify new inhibitors for drug development [11]. For molecular docking between receptor and ligand used AUTODOCK and RACCOON tools. The ADMET studies done using ilab.acdlabs.com online tool for targeted compound. That studies shows that compounds could be exploited as oral drug candidate [12].
Material and Methodology
Target selection
Innate immunity plays an important role in defending host from invading pathogens. IFNs are induce to antiviral response that can defend host from invading pathogen. From literature we found that when human bocavirus attack on the host then VP2 protein of HBoV inhibit the function of Retinoic Acid– Inducible Gene-I (RIG-1), that RIG-1 protein activate the IFNs promoters [13]. In addition, from UNIPROT database [14] found that VP2 protein helps in making of capsid of virus. Therefore, we have targeted Human bocavirus VP2 protein to design a novel drug. Human bocavirus belongs to parvoviridae family. VP2 protein sequence obtained from NCBI in FASTA format of 542 amino acid long, and accession number was AGL08709.1 [15].
Protein structure prediction
Protein structure prediction of Human Bocavirus VP2 protein was done with the assistance of Modeller tool to understand its physicochemical properties. Modeller is most commonly used tool for prediction of protein structure. It supports us to find the 3D structure of protein [16]. Thus, it helps us to study the properties of the protein structure. We have use the Max Planck Modeller (https://toolkit.tuebingen.mpg.de/modeller) [17] to design a 3D structure of target (Figure 1).

Figure 1: 3D structure of human bocavirus VP2 protein by Max Planck Modeller.
Pocket detection: To perform docking on any target molecule, it is very important to recognise active site of that molecule. Sites of activity in proteins generally lie in cavities. The shape and size of protein cavities control the three-dimensional geometry of ligands that can strongly bind there; i.e., they must fit proper similar to a hand in a glove. Therefore, a minimal requirement for drug activity is that the molecule sterically fit the area of hidden volume carving the active site cavity, through approximately allowance for induced fit [18].
There are several commonly used methodologies including which our used SiteHound-web tool. The SiteHound algorithm finds potential ligand binding sites by identifying regions characterized by promising non-bonded interactions with a chemical probe [19]. Presently, a ‘Carbon’ probe and a ‘Phosphate’ probe are available for the identification of binding sites for drug-like molecules, and ligands containing phosphate groups, separately [20]. SiteHound can help to find to potential binding site where drug can bind.
Molecular docking
The mechanism of binding of drug with the target protein is called docking [21]. The docking can be used to find inhibitors for specific target proteins and thus to design new stable drugs from docking results [22]. Docking can calculated by binding energy. In this project, we have used AutoDcok software for docking. AutoDock is a suite of free open-source software for the computational docking and virtual screening of small molecules to macromolecular receptors. The suite currently includes several complementary tools: in which we have used AutoDock (a computational docking program based on an empirical free-energy force field and rapid Lamarckian genetic algorithm search method [23]) and Raccoon (an interactive graphical tool for virtual screening and analysis [11]) tool. We used Human bocavirus VP2 protein as a receptor. Small molecules and zinc database molecules as ligands.
Pharmacophore model preparation
Pharmacophore designing is the early step before starting the screening. The pharmacophore modal is created using our LigandScout software [8]. LigandScout software can assist to screen out molecule from the pharmacophore model. Virtual screening recognized as one of the most important computational technique used to separate from unwanted molecules within compound libraries, which has done as early as possible in order to reduce drug discovery costs. This method simply can built into an automatic workflow environment. LigandScout software tool that allows to fast and transparently deriving Ligand-based pharmacophores from structural data of macromolecule/ligand complexes in a fully automated and appropriate way. LigandScout starts with a macromolecule/ligand complex and automatically detects bound ligands generating a standard residue around the non-standard residues [24].
For Designing a Pharmacophore model, use the best three small molecules on the bases of binding energy with target. Ligand-ligand pharmacophore used to make pharmacophore model. Further generating the pharmacophore model this same tool used to perform virtual screening of the pharmacophore against already created ligand library of approximately nine lakh ligand molecules each obtained from ZINC database. We got 22010 ligands from virtual screening again these ligands subjected to docking with Human bocavirus VP2 protein. From that, the ligands with the best minimum binding energies selected for further studies.
ADME-Tox analysis: After docking ADMET analysis and toxicity prediction done. An important step remains in the drug discovery method, mostly in the advanced stages of lead discovery, is analysis of the ADME and over toxicity properties of drug candidates. Over 50% of the molecules unsuccessful due to ADMET deficiencies during development. To evade this failure at the development, a set of in vitro ADME screens has been implemented in most pharmaceutical companies with the purpose of removal compounds in the discovery stage that are likely to fail further down the line. PreADME is suitable for high throughput screening and combinatorial chemistry library design considering the Linpinski’s rule or lead-like rule, drug absorption and water solubility [25]. We used the PreADMET (http:// preadmet.bmdrc.kr/) tool [26]. The PreADMET program provides rapid and reliable data of drug-likeness and ADME properties [27].
Result
Human bocavirus 1 vp2 protein 3D structure was not available on PDB database [28]. Than vp2 protein sequence retrieve from NCBI [29]. Than to design, a 3D structure of protein used Max Planck modeller online tool (https:// toolkit.tuebingen.mpg.de/modeller) [17]. 3D structure of protein showing into Figure 1.
To perform docking on any target molecule, it is very important to recognise active site of that molecule, where ligands will attach. We found out the active site of the human bocavirus VP2 protein by the help of SiteHound-web tool. the active sites are as follows GLN 101, SER 103, GLU 174, ASP 175, VAL 176, MET 177, PRO 178, ASP179, LEU 180, LYS 153, THR 184, TRP 185, LYS 186, GLN 361, THR 362, PRO 363, GLU 507, TYR 522, GLN 524. Further finding the active site of Human bocavirus VP2 protein molecular docking performed by total 126 small molecules as ligands and Human bocavirus 1 VP2 protein as target (Table 1 and Figure 2).
| Sr. no | Small molecule ligands | Binding energies |
|---|---|---|
| 1 | ZINC64624173 | -7.99 |
| 2 | ZINC4099018 | -7.88 |
| 3 | ZINC64624174 | -7.87 |
| 4 | ZINC85537026 | -7.75 |
| 5 | ZINC18210358 | -7.70 |
| 6 | ZINC49627537 | -7.44 |
| 7 | ZINC59789263 | -7.35 |
| 8 | ZINC49052497 | -7.33 |
| 9 | ZINC9574816 | -7.33 |
| 10 | ZINC95754342 | -7.21 |
Table 1: Human bocavirus VP2 protein molecular docking top result.

Figure 2: Structure of ligand (a) Zinc64624173, (b) Zinc64624174 (c) Zinc04099018 and (d) 3D structure of the pharmacophore molecule created with help of best three ligands.
The first three ligand molecules having the most minimum binding energies given bellow were used to create pharmacophore model. Before creating pharmacophore model minimize the molecule energy through LigandScout software. Than pharmacophore model created through first three molecule ZINC64624173 (OLEANANE), ZINC4099018 (RHODAMINE 123) and ZINC64624174 (HOPANE). Total 9- lac molecules library created from zinc database. This library screen out through pharmacophore model by LigandScout software. Total 22010 ligands library created. We performed a molecular docking of all 22010 ligands with VP2 protein. Finally, we got the result showing 10 best ligands molecules on basis of binding energy. If campare between both result of small molecules and molecules get from pharmacophore modelling than result from pharmacophore modelling was better. From small molecules got lowest binding energy was -7.99 (ZINC64624173) (Table 1) and molecules from pharmacophore models gets lowest energy -10.29(ZINC72330347) (Table 2) which is showing much more higher energy comparative small molecules that mean that molecule will show higher stability. The ligands we got form the pharmacophore screening shows the best binding energies, hence are used for the further studies.
| Sr.no | ZINC ID | Chemical structure | Binding energy |
|---|---|---|---|
| 1 | zinc72330347 | 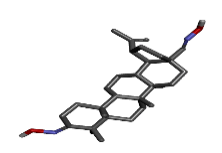 |
-10.29 |
| 2 | Zinc12606400 | 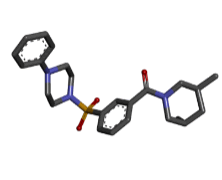 |
-9.92 |
| 3 | Zinc72330345 | 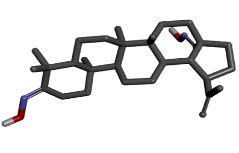 |
-9.89 |
| 4 | Zinc15670620 | 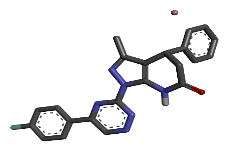 |
-9.66 |
| 5 |
Zinc72330338 | 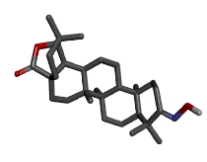 |
-9.36 |
| 6 |
Zinc23078235 | 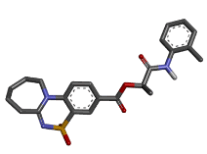 |
-9.34 |
| 7 |
Zinc04015296 | 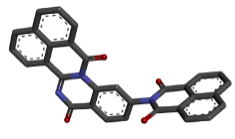 |
-9.30 |
| 8 |
Zinc04702789 | 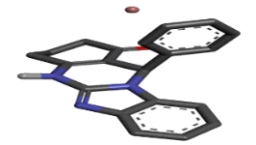 |
-9.18 |
| 9 | Zinc02964193 | 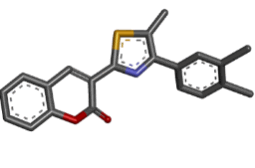 |
-9.16 |
| 10 | Zinc72330341 | 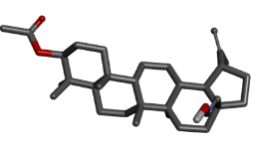 |
-9.04 |
Table 2: Best ten ligands were selected for ADME-Tox Analysis.
Analysis of 10 molecules compounds from pharmacophore model result (Tables 3A and 3B) predicted about ADMET (Adsorption, Distribution, Metabolism, Excretion and Toxicity) through Pre-ADMET software [27]. ADME-Tox analysis was performed on the above ligands was done with the help of online tool Pre-Admet (https://preadmet.bmdrc.kr/) [26] and following results were obtained.
| Sno. | ZINC ID’S | ADME PROPERTIES | ||||
|---|---|---|---|---|---|---|
| BBB | HIA | CACO-2 | Rule of 5 | MDDR | ||
| 1 | zinc72330347 | 8.9202 | 94.171 | 21.276 | Suitable | Mid-structure |
| 2 | zinc12606400 | 2.497 | 97.487 | 21.71 | Suitable | Mid-structure |
| 3 | zinc72330345 | 8.9208 | 94.1717 | 21.276 | Suitable | Mid-structure |
| 4 | zinc15670620 | 2.3804 | 98.772 | 22.319 | Suitable | Mid-structure |
| 5 | zinc72330338 | 7.98312 | 95.77 | 23.19 | Suitable | Mid-structure |
| 6 | zinc23078235 | 0.0379 | 91.51 | 13.662 | Failed | Mid-structure |
| 7 | zinc04015296 | 0.9889 | 0.9815 | 0.6252 | - | - |
| 8 | zinc04702789 | 3.818 | 97.54 | 35.31 | Suitable | Mid-structure |
| 9 | zinc02964193 | 0.949 | 1.001 | 0.5497 | - | - |
| 10 | zinc723303541 | 9.926 | 96.08 | 26.53 | Suitable | Mid-structure |
Table 3A: ADME analysis of best dock molecules.
| Sno. | ZINC ID’S | TOXICITY | |||
|---|---|---|---|---|---|
| WDI | AMES | Rat | Mouse | ||
| 1 | zinc72330347 | Out of 90% cutoff | Mutagen | Positive | Positive |
| 2 | zinc12606400 | Out of 90% cutoff | Mutagen | Negative | Negative |
| 3 | zinc72330345 | Out of 90% cutoff | Mutagen | Positive | Positive |
| 4 | zinc15670620 | Failed | Non-mutagenic | - | - |
| 5 | zinc72330338 | Out of 90% cutoff | Non-mutagenic | Positive | Negative |
| 6 | zinc23078235 | Failed | Mutagen | Negative | Negative |
| 7 | zinc04015296 | - | Non ames toxic | Non- carcinogenic | - |
| 8 | zinc04702789 | Failed | Mutagen | Out of range | - |
| 9 | zinc02964193 | - | - | Non-carcinogenic | - |
| 10 | zinc723303541 | Out of 90% cutoff | Mutagen | Positive | Positive |
Table 3B: Toxicity analysis of best dock molecules.
In the following result, we studied their ADME properties, drug likeliness and toxicity. All The ligand molecules displayed high absorption in central nervous system except for ZINC23078235 showed minimum absorption in the central nervous system. All the molecules show high human intestinal absorption except ZINC04015296 and low permeability was seen in CaCO-2 test of ZINC04015296 and ZINC02964193 show low permeability. In druglikeliness ZINC23078235 molecules fail to follow Lipinski rule of 5. All ligand molecules are in mid-structure range. In toxicity test ZINC15670620, ZINC23078235, ZINC04702789 ligand molecules failed in the WDI (World Drug Index) and in AMES test ZINC15670620, ZINC72330338 ZINC04015296 are not mutagenic. The ligand molecule ZINC72330347, ZINC72330345, ZINC04015296, ZINC02964193, ZINC72330341 displayed non-carcinogenicity in Rat and Mouse respectively. Considering the ADMET results and minimum binding energies, we can consider ZINC72330347, ZINC12606400, ZINC72330345, ZINC72330338 and ZINC723303541 to be investigational new drug candidate for treatment infection caused by Human bocavirus 1. However, after in wet analysis can say that which molecule out of 10 can be used as a drug molecules.
Conclusion
This study shows that targeting VP2 protein can effect Human bocavirus 1 persistence. In this research paper, we used small molecules as ligand and VP2 protein as a receptor. From best small molecule pharmacophore model created from screening of molecule. Commencing that model total 9 lac molecules are screen out finally 22010 molecules left which used as ligand and docking perform with VP2 protein. While comparisons are done than found, that molecule from pharmacophore modelling give best result. From small molecule we got minimum binding energy was -7.99 (ZINC64624173) (Table 1) and molecules from pharmacophore models gets lowest energy -10.29(ZINC72330347) (Table 1). Further, when ADMET analysis was done of top minimum binding energy molecules only 5 molecules (ZINC72330347, ZINC12606400, ZINC72330345, ZINC72330338 and ZINC723303541) pass ADMET test. These molecules could be used as inhibitor for VP2 protein. VP2 protein of HBoV inhibited the function of Retinoic Acid–Inducible Gene-I (RIG-1), that RIG-1 protein activate the IFNs promoters in host. In addition, from UNIPROT database found that VP2 protein helps in making of capsid of virus. If these molecules inhibit the function of VP2 protein than virus might not be, inhibit the function of RIG- 1 gene. Therefore, host immunity not can bout with virus. In addition, virus capsid will effect that can make virus weak. Finally if they pass clinical train than they could be used as drug against for Human Bocavirus 1.
References
- Allander T, Tammi MT, Eriksson M, Bjerkner A, Tiveljung-Lindell A, Andersson B. Cloning of a human parvovirus by molecular screening of respiratory tract samples. Proc Natl Acad Sciences U S A 2005; 102: 12891-12896.
- Jartti T, Hedman K, Jartti L, Ruuskanen O, Allander T. Human bocavirus-the first 5 years. Rev Med Virol 2012; 22: 46-64.
- Milder E, Arnold JC. Human metapneumovirus and human bocavirus in children. Pediatr Res 2009; 65: 78R-83R.
- Kantola K, Hedman L, Allander T, Jartti T, Lehtinen P, Ruuskanen O. Serodiagnosis of human bocavirus infection. Clin Infect Dis 2008; 46: 540-546.
- Macielag MJ. Chemical properties of antimicrobials and their uniqueness. Antibiotic Discovery and Development: Springer, Berlin, 2012.
- Samanen J. Introduction to Biological and Small Molecule Drug Research and Development: Chapter 5. Similarities and differences in the discovery and use of biopharmaceuticals and small-molecule chemotherapeutics: Elsevier Inc. Chapters; 2013.
- Wolber G, Langer T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J Chem Inf Model 2005; 45: 160-169.
- Wolber G, Dornhofer AA, Langer T. Efficient overlay of small organic molecules using 3D pharmacophores. J Comput Aided Mol Design 2006; 20: 773-788.
- Wolber G, Seidel T, Bendix F, Langer T. Molecule-pharmacophore superpositioning and pattern matching in computational drug design. Drug Disc Today 2008; 13: 23-29.
- Bröer BM, Gurrath M, Höltje H-D. Molecular modelling studies on the ORL1-receptor and ORL1-agonists. J Comput Aided Mol Design 2003; 17: 739-754
- Forli S, Huey R, Pique ME, Sanner MF, Goodsell DS, Olson AJ. Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nature Protocols 2016; 11: 905-919.
- Trifunovic J, Borcic V, Vukmirovic S, Kon SG, Mikov M. Retention data of bile acids and their oxo derivatives in characterization of pharmacokinetic properties and in silico ADME modeling. Euro J Pharmaceut Sci 2016; 92: 194-202.
- Zhang Z, Zheng Z, Luo H, Meng J, Li H, Li Q. Human bocavirus NP1 inhibits IFN-ß production by blocking association of IFN regulatory factor 3 with IFNB promoter. J Immunol 2012; 189: 1144-1153
- Consortium U. The universal protein resource (UniProt). Nucleic Acids Res 2008; 36: D190-D5.
- Wheeler DL, Barrett T, Benson DA, Bryant SH, Canese K, Chetvernin V. Database resources of the national center for biotechnology information. Nucleic Acids Res 2007; 35: D5-D12.
- Eswar N, Eramian D, Webb B, Shen MY, Sali A. Protein structure modeling with MODELLER. Methods Mol Biol 2008; 426: 145-159.
- Šali A, Potterton L, Yuan F, van Vlijmen H, Karplus M. Evaluation of comparative protein modeling by MODELLER. Proteins 1995; 23: 318-326.
- Brady GP Jr, Stouten PF. Fast prediction and visualization of protein binding pockets with PASS. J Comput Aided Mol Des 2000; 14: 383-401.
- Ghersi D, Sanchez R. Improving accuracy and efficiency of blind protein-ligand docking by focusing on predicted binding sites. Proteins 2009; 74: 417-424.
- Hernandez M, Ghersi D, Sanchez R. SITEHOUND-web: a server for ligand binding site identification in protein structures. Nucleic Acids Res 2009; 37: W413-416.
- Moitessier N, Englebienne P, Lee D, Lawandi J, Corbeil CR. Towards the development of universal, fast and highly accurate docking/scoring methods: a long way to go. Br J Pharmacol 2008; 153: S7-S26.
- Talukdar M. Stable Drug Designing by Minimizing Drug-Protein Interaction Energy using PSO: Jadavpur University; 2014.
- Goodsell DS, Olson AJ. Automated docking of substrates to proteins by simulated annealing. Proteins 1990; 8: 195-202.
- Steindl TM, Schuster D, Wolber G, Laggner C, Langer T. High-throughput structure-based pharmacophore modelling as a basis for successful parallel virtual screening. J Comput Aided Mol Design 2006; 20: 703-715.
- Seal A, Aykkal R, Babu RO, Ghosh M. Docking study of HIV-1 reverse transcriptase with phytochemicals. Bioinformation 2011; 5: 430-439.
- BMDRC P. 2.0, Bmdrc, Seoul. Korea. 2007.
- Singh A, Jatav VK, Sharma S. Virtual screening and admet analysis for identification of inhibitors against acetylcholinesterase associated with alzheimer's disease. Int J Pharmacy Pharmaceut Sci 2014; 6: 155-159.
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN. The Protein Data Bank. Nucleic Acids Res 2000; 28: 235-242.
- Stojmirovic A, Yu YK. Information flow in interaction networks. J Comput Biol 2007; 14: 1115-1143.